Kit de biblioteca TIANSeq DirectFast (illumina)
Recursos
■ Boa uniformidade de sequenciamento: Sem polarização de base do processo de fragmentação do DNA e do processo de amplificação por PCR.
■ Alta eficiência de conversão de biblioteca: a construção de biblioteca de alta eficiência pode ser garantida para amostras de 1 ng de DNA.
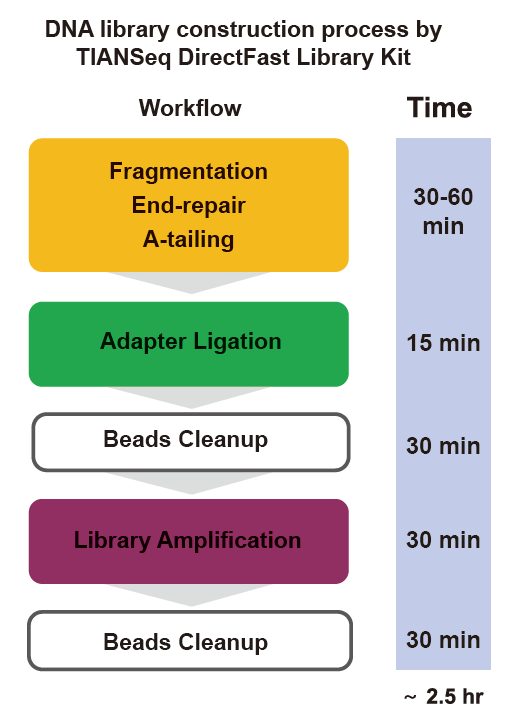
■ Operação rápida: todo o processo de construção da biblioteca precisa de apenas 2,5 horas.
■ Custo-benefício: Não são necessários instrumentos e equipamentos especiais。
Especificação
Modelo: Preparação de biblioteca de DNA para plataforma de sequenciamento de alto rendimento Illumina
Amostra: DNA genômico ou DNA de fragmento grande
Alvo: DNA de fita dupla
Iniciando a entrada de amostra: 1 ng- 1 μg
Tempo de operação: 2,5 horas
Aplicativos downstream: Sequenciamento na plataforma Illumina
Todos os produtos podem ser personalizados para ODM / OEM. Para detalhes,clique em Serviço personalizado (ODM / OEM)




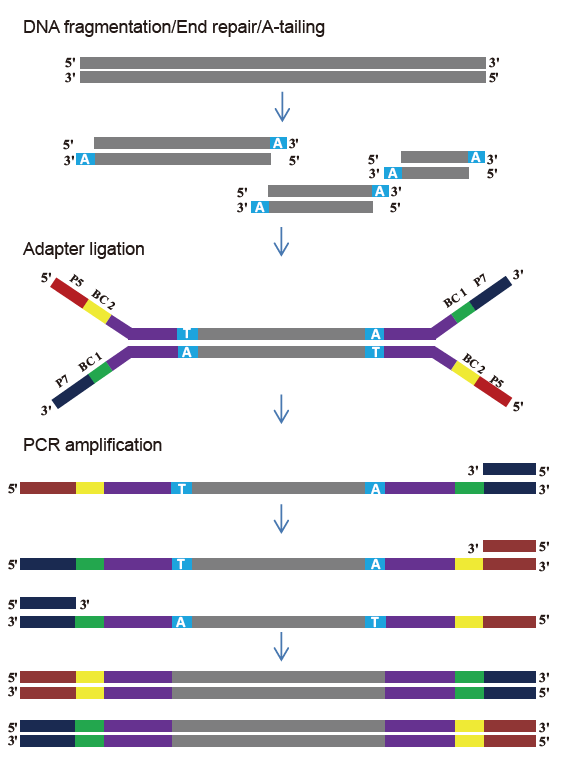
Entrada de amostra flexível e tamanho fragmentado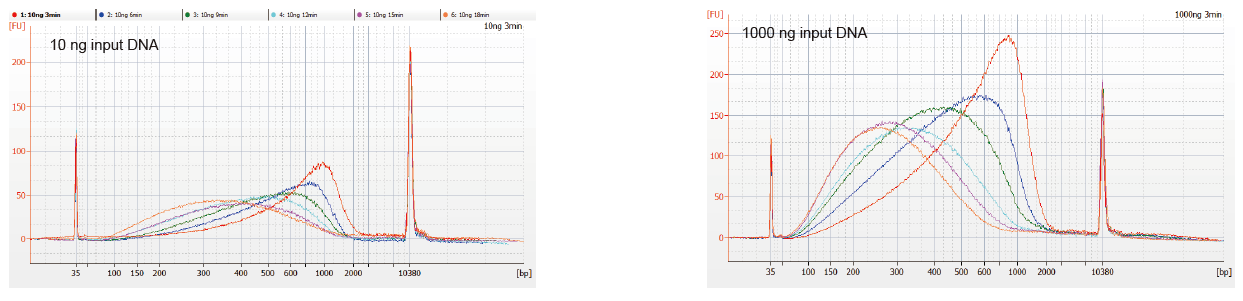 |
Figura 1. Perfis de fragmentação de DNA de diferentes tempos de reação. 10 ng e 1000 ng de DNA foram fragmentados usando o kit TIANSeq DirectFast DNA Library. Os produtos de reação tratados com diferentes tempos de reação foram purificados por esferas magnéticas Ampure XP de 1,8 × e analisados por Angilent 2100. |
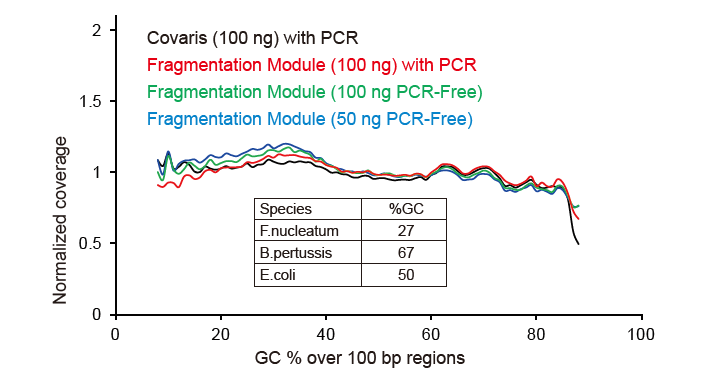
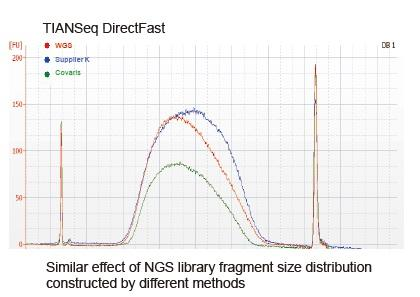
Cobertura de sequenciamento semelhante a Covaris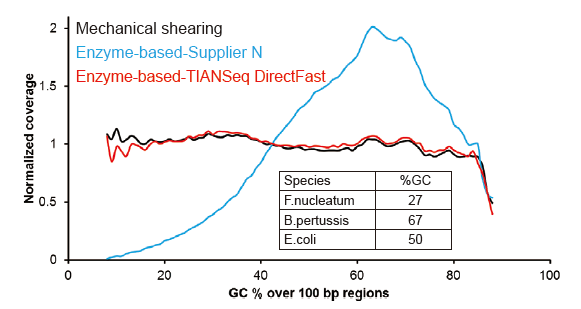 |
Figura 2. Comparação da cobertura do genoma de diferentes métodos de preparação de biblioteca. Três DNA genômico bacteriano com diferentes conteúdos de GC são equimolares mistos, e o resultado da cobertura do genoma de sequenciamento de 100 ng de bibliotecas de DNA misto usando esses métodos foram comparados. Os resultados mostram que o TIANSeq DirectFast Library Kit tem o mesmo efeito sobre a fragmentação do DNA que o cisalhamento mecânico, e não há polarização de base para a fragmentação. |
Sem viés sistemático para DNA de entrada tão baixo quanto 1 ng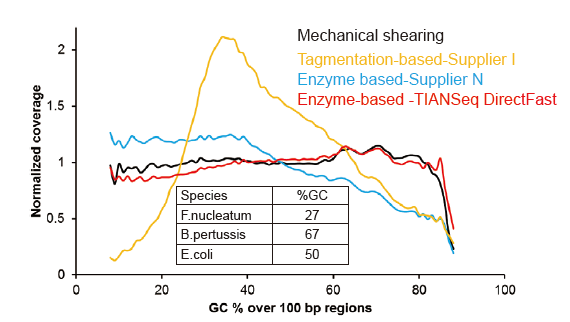 |
Figura 3. Comparação da cobertura do genoma de diferentes métodos de preparação de biblioteca. Três DNA genômico bacteriano com diferentes conteúdos de GC são equimolares mistos, e o resultado da cobertura do genoma de sequenciamento de 1 ng de bibliotecas de DNA misto usando esses métodos foram comparados. Os resultados mostram que o TIANSeq DirectFast Library Kit tem efeito de fragmentação consistente com o cisalhamento mecânico, mesmo para entrada de DNA tão baixo quanto 1 ng, e não há polarização de base. |
| Capaz de fluxo de trabalho livre de PCR
|
Figura 4. Diferentes entradas de DNA genômico foram usadas para construir a biblioteca por PCR ou construção de biblioteca livre de PCR, e os resultados de cobertura do genoma foram comparados. Os resultados mostram que com a operação de um tubo e as etapas eficientes de construção da biblioteca, a biblioteca de DNA construída com o kit TIANSeq DirectFast Library mantém uma alta consistência com o cisalhamento mecânico na distribuição da cobertura da sequência de fragmentos para o fluxo de trabalho livre de PCR de enriquecimento de PCR. |
Estatísticas de eficiência e rendimento de construção de bibliotecas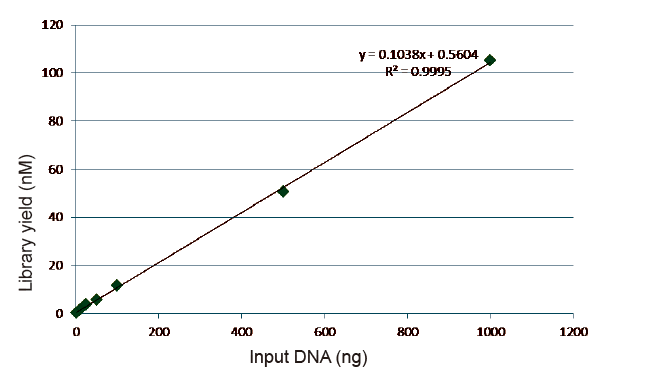 |
Figura 5. Resultados da análise quantitativa do DNA da biblioteca obtido por qPCR após a construção da biblioteca pelo método livre de PCR para amostras com diferentes quantidades iniciais (1, 10, 25, 50, 100, 500, 1.000 ng). A análise de regressão linear mostra que o rendimento da biblioteca tem um bom relacionamento linear em uma ampla faixa de entrada de amostra. Para entrada de DNA tão baixa quanto 1 ng, a eficiência da construção da biblioteca não diminui. |
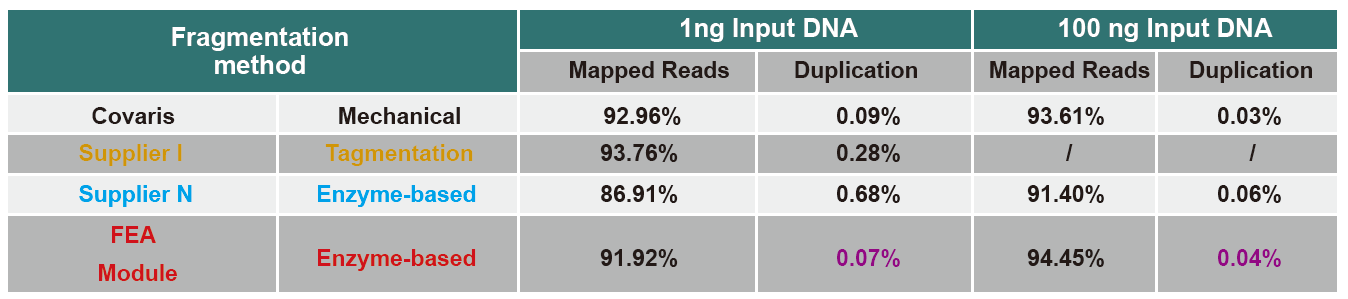
Comparação de dados de sequenciamento de produtos diferentes
Atualmente, a tecnologia de sequenciamento de alto rendimento é baseada principalmente na tecnologia de sequenciamento de próxima geração. Como o comprimento de leitura da tecnologia de sequenciamento de próxima geração é limitado, devemos quebrar a sequência de comprimento total em bibliotecas de pequenos fragmentos para sequenciar. De acordo com as necessidades de diferentes experimentos de sequenciamento, geralmente escolhemos sequenciamento de extremidade única ou sequenciamento de extremidade dupla. Atualmente, os fragmentos de DNA da biblioteca de sequenciamento de próxima geração são geralmente distribuídos na faixa de 200-800 pb.
a) O DNA é de baixa qualidade e contém inibidores. Use amostras de DNA de alta qualidade para evitar a inibição da atividade enzimática.
b) A quantidade de amostra de DNA é insuficiente ao usar o método livre de PCR para construir a biblioteca de DNA. Quando a entrada do DNA fragmentado excede 50 ng, o fluxo de trabalho livre de PCR pode ser realizado seletivamente durante o processo de construção da biblioteca. Se o número de cópias da biblioteca for muito baixo para ser sequenciado diretamente, a biblioteca de DNA pode ser amplificada por PCR após a ligação do adaptador.
c) A contaminação de RNA leva a uma quantificação de DNA inicial imprecisa A contaminação de RNA pode existir no processo de purificação do DNA genômico, o que pode levar à quantificação de DNA imprecisa e carregamento insuficiente de DNA durante a construção da biblioteca. O RNA pode ser removido por tratamento com RNase.
A-1
a) Fragmentos pequenos (60 bp-120 bp) aparecem Fragmentos pequenos geralmente são fragmentos adaptadores ou dímeros formados por adaptadores. A purificação com esferas magnéticas Agencourt AMPure XP pode efetivamente remover esses fragmentos do adaptador e garantir a qualidade do sequenciamento.
b) Fragmentos grandes aparecem na biblioteca após a amplificação por PCR O tamanho do fragmento de DNA da biblioteca aumentará em 120 pb após o adaptador ser ligado. Se o fragmento de DNA aumentar em mais de 120 bp após a ligação do adaptador, isso pode ser causado pela amplificação anormal do fragmento ou pela amplificação PCR excessiva. A redução do número de ciclos de PCR pode evitar a situação.
c) Tamanho anormal dos fragmentos de DNA da biblioteca após a ligação do adaptador O comprimento do adaptador neste kit é 60 bp. Quando as duas extremidades do fragmento são ligadas aos adaptadores, o comprimento aumentará apenas 120 bp. Ao usar um adaptador diferente do fornecido por este kit, entre em contato com o fornecedor para fornecer informações relevantes, como o comprimento do adaptador. Certifique-se de que o fluxo de trabalho e a operação do experimento sigam as etapas descritas no manual.
d) Tamanho anormal do fragmento de DNA antes da ligação do adaptador A razão para este problema pode ser causada por condições de reação incorretas durante a fragmentação do DNA. Diferentes tempos de reação devem ser usados para diferentes entradas de DNA. Se a entrada de DNA for superior a 10 ng, recomendamos escolher o tempo de reação de 12 min como o tempo de início para a otimização, e o tamanho do fragmento produzido neste momento está principalmente na faixa de 300-500 bp. Os usuários podem aumentar ou diminuir o comprimento dos fragmentos de DNA por 2 a 4 minutos de acordo com seus próprios requisitos para otimizar os fragmentos de DNA com o tamanho necessário.
A-2
a) O tempo de fragmentação não é otimizado Se o DNA fragmentado for muito pequeno ou muito grande, consulte as Diretrizes para Seleção do Tempo de Fragmentação fornecidas na instrução para determinar o tempo de reação e usar este ponto de tempo como um controle, adicionalmente configurar um sistema de reação para prolongar ou encurtar 3 min para fazer um ajuste mais preciso no tempo de fragmentação.
A-3
Distribuição de tamanho anormal de DNA após o tratamento de fragmentação
a) Método de descongelamento incorreto do reagente de fragmentação ou o reagente não está completamente misturado após o descongelamento. Descongele o reagente 5 × Fragmentation Enzyme Mix no gelo. Depois de descongelado, misture o reagente uniformemente sacudindo suavemente o fundo do tubo. Não vortex o reagente!
b) A amostra de DNA de entrada contém EDTA ou outros poluentes. O esgotamento dos íons de sal e agentes quelantes na etapa de purificação do DNA é particularmente importante para o sucesso do experimento. Se o DNA for dissolvido em 1 × TE, use o método fornecido na instrução para realizar a fragmentação. Se a concentração de EDTA na solução for incerta, recomenda-se purificar o DNA e dissolvê-lo em água desionizada para a reação subsequente.
c) Quantificação de DNA inicial imprecisa O tamanho do DNA fragmentado está intimamente relacionado à quantidade de DNA de entrada. Antes do tratamento de fragmentação, a quantificação precisa do DNA usando Qubit, Picogreen e outros métodos é essencial para determinar a quantidade exata de DNA no sistema de reação.
d) A preparação do sistema de reação não segue as instruções A preparação do sistema de reação fragmentado deve ser realizada em gelo estritamente de acordo com as instruções. Para garantir o melhor efeito, todos os componentes da reação devem ser colocados em gelo e a preparação do sistema de reação deve ser realizada após o resfriamento completo. Depois de concluída a preparação, agite ou pipete para misturar bem. Não vortex!
1. O método de mistura impróprio (vórtice, oscilação violenta, etc.) causará distribuição anormal dos fragmentos da biblioteca (como mostrado na figura a seguir), afetando assim a qualidade da biblioteca. Portanto, ao preparar a solução de reação da Mistura de Fragmentação, pipete suavemente para cima e para baixo para misturar ou use a ponta do dedo para agitar e misturar uniformemente. Tenha cuidado para não misturar com o vórtice.
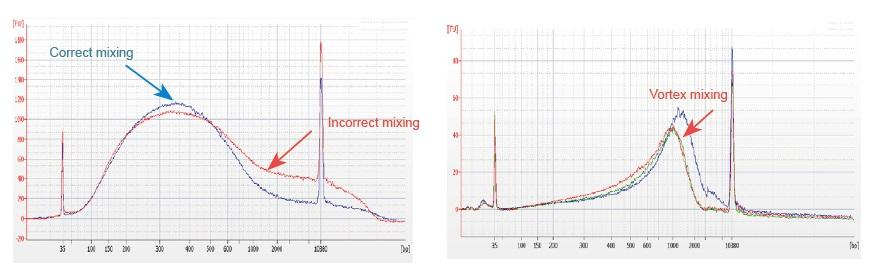
2. O DNA de alta pureza deve ser usado para a construção da biblioteca
■ Boa integridade de DNA: A banda de eletroforese é superior a 30 kb, sem cauda
■ OD260 / 230:> 1,5
■ OD260 / 280: 1,7-1,9
3. A quantidade de DNA de entrada deve ser precisa. Sugere-se usar os métodos Qubit e PicoGreen para quantificar o DNA, em vez de Nanodrop.
4. O conteúdo de EDTA na solução de DNA deve ser determinado. O EDTA tem uma grande influência na reação de fragmentação. Se o conteúdo de EDTA for alto, a purificação do DNA precisa ser realizada antes do teste subsequente.
5. A solução da reação de fragmentação deve ser preparada em gelo. O processo de fragmentação é sensível à temperatura e ao tempo de reação (especialmente após a adição do intensificador). Para garantir a precisão do tempo de reação, prepare o sistema de reação no gelo.
6. O tempo de reação de fragmentação deve ser preciso O tempo de reação da etapa de fragmentação afetará diretamente o tamanho dos produtos do fragmento, afetando assim a distribuição do tamanho dos fragmentos de DNA na biblioteca.
1. Que tipo de amostra é aplicável a este kit?
O tipo de amostra aplicável deste kit pode ser RNA total ou mRNA purificado com boa integridade de RNA. Se o RNA total for usado para construir a biblioteca, é recomendado usar o kit de depleção de rRNA (Cat # 4992363/4992364/4992391) para remover o rRNA primeiro.
2. As amostras FFPE podem ser usadas para construir a biblioteca com este kit?
O mRNA em amostras FFPE será degradado até certo ponto, com integridade relativa pobre. Ao utilizar este kit para a construção da biblioteca, recomenda-se otimizar o tempo de fragmentação (encurtar o tempo de fragmentação ou não realizar a fragmentação).
3. Usando a etapa de seleção de tamanho fornecida no manual do produto, o que pode fazer com que o segmento inserido apareça um ligeiro desvio?
A seleção do tamanho deve ser realizada estritamente de acordo com a etapa de seleção do tamanho neste manual do produto. Se houver desvio, pode ser que as esferas magnéticas não estejam balanceadas para a temperatura ambiente ou não estejam totalmente misturadas, a pipeta não seja precisa ou o líquido permaneça na ponta. Recomenda-se usar as pontas com baixa adsorção para o experimento.
4. Seleção de adaptadores na construção da biblioteca
O kit de construção da biblioteca não contém reagente adaptador e é recomendável usar este kit junto com o Adaptador de índice único TIANSeq (Illumina) (4992641/4992642/4992378).
5. QC da biblioteca
Detecção quantitativa da biblioteca: Qubit e qPCR são usados para determinar a concentração de massa e a concentração molar da biblioteca, respectivamente. A operação está estritamente de acordo com o manual do produto. A concentração da biblioteca geralmente atenderá aos requisitos de sequenciamento NGS. Detecção da faixa de distribuição da biblioteca: Uso do Agilent 2100 Bioanalyzer para detectar a faixa de distribuição da biblioteca.
6. Seleção do número do ciclo de amplificação
De acordo com as instruções, o número de ciclos de PCR é 6-12 e o número de ciclos de PCR necessários deve ser selecionado de acordo com a entrada da amostra. Em bibliotecas de alto rendimento, a superamplificação geralmente ocorre em vários graus, o que se manifesta por um pico ligeiramente maior após o pico da faixa alvo na detecção do Agilent 2100 Bioanalyzer, ou a concentração detectada de Qubit é menor do que a de qPCR. A superamplificação leve é um fenômeno normal, que não afeta o sequenciamento da biblioteca e a análise de dados subsequente.
7. Picos aparecem no perfil de detecção do Agilent 2100 Bioanalyzer
O aparecimento de picos na detecção do Agilent 2100 Bioanalyzer se deve à fragmentação desigual das amostras, onde haverá mais fragmentos em determinado tamanho, e isso se tornará mais óbvio após o enriquecimento por PCR. Nesse caso, sugere-se não realizar a seleção do tamanho, ou seja, definir a condição de fragmentação para 94 ° C por 15 min incubado, onde a distribuição do fragmento é pequena e concentrada, e a homogeneidade pode ser melhorada.
Categorias de produtos
PORQUE ESCOLHER-NOS
Desde a sua criação, nossa fábrica tem desenvolvido produtos de primeira classe com a adesão ao princípio
de qualidade primeiro. Nossos produtos ganharam excelente reputação na indústria e são valiosos e confiáveis entre clientes novos e antigos.
- Tel: +86 010-59822688
- Edifício 5, nº 86, Shuangying West Road, distrito de Changping, Pequim.
- people@tiangen.com



